



Что такое синдром Барттера?

Что такое синдром Барттера?
Синдром псевдо — Бартерра
Опубликовано Ольга Алекина в 15.10.2012 15.10.2012
Bartter Frederic Crosby, американский эндокринолог.
Тяжелое осложнение муковисцидоза — синдром псевдо-Бартера, которое может развиваться у больных в экстремальных погодных условиях при несоблюдении рекомендаций по дополнительному обеспечению солями натрия и хлора и коррекции водной нагрузки.
Впервые признаки, хаpaктерные для псевдо-Барттер синдрома, описали в 1951 г Kessler WR, Andersen DH. Авторы наблюдали «тепловую прострацию» у детей жарким летом 1948 года, и у большинства из этих детей был муковисцидоз. Авторы исследовали причины высокой чувствительности детей с муковисцидозом к теплу, и обнаружили аномально высокое содержание натрия и хлора в поте – то есть нетипичное для здоровых людей повышенное выделение солей. Это наблюдение было важным шагом в понимании патогенеза муковисцидоза.
В 1959 г в Австралии, в стране с жарким климатом, Douglas WAC наблюдал двоих детей с симптомами острого недостатка солей. Детям рекомендовали увеличить потрeбление поваренной соли до 4 грамм в день в жаркий период, и состояние детей нормализовалось.
В 1971 г Gottlieb RP., а в 1979 Beckerman RC и Taussig LM. описали случаи метаболического алкалоза, гипохлоремии и гипокалиемии у детей с муковисцидозом, не связанные с выраженным обезвоживанием и перегревом. Это осложнение стали назвать синдромом псевдо-Баттера.
Синдром псевдо-Барттера — симптомокомплекс, хаpaктеризующийся гипокалиемией, гипохлоремией, метаболическим алкалозом, обезвоживанием, повышенной активностью ренина плазмы крови, повышенным содержанием альдостерона в крови.
Причины развития синдрома псевдо-Баттера
Неправильная работа (или его отсутствие) белка – регулятора натриевого обмена в клетках приводит к изменению водно-солевого баланса в организме. У пациентов в условиях жаркого климата, а также у младенцев с еще не совершенной системой терморегуляции, с повышенным выделением натрия с потом (соленый вкус кожи), концентрация натрия и других солей в крови снижается. У детей, находящихся на грудном вскармливании, особенно живущих в жарком климате, риск развития этого синдрома особенно высок связи с низким содержанием солей в грудном молоке.
Значение Калия, Натрия и Хлора в организме
Калий (К+) создает и поддерживает электрический мембранный потенциал клеток. Участвует в регуляции осмотического давления, метаболизма глюкозы и белков. Важную роль играет в проведении нервных импульсов и в формировании потенциала действия нервных и мышечных клеток. Участвует в иммунных процессах.
Концентрация калия в плазме (сыворотке) зависит от равновесия следующих процессов: поступления калия извне, распределения в организме и выведения (почками, потовыми железами, через кишечник и т. п.). Запасов калия в организме не существует. Поэтому даже при небольших изменениях концентрации калия внутри клеток, значительно изменяется его концентрация в плазме. Потрeбление калия клетками стимулируется инсулином (гормоном поджелудочной железы), также захват калия клетками усиливается под действием катехоламинов, альдостерона (гормонов надпочечников). Изменения рН (кислотности) крови приводят к изменению содержания К+ в клетках: при ацидозе — он выходит из клеток в плазму, при алкалозе поступает внутрь клеток. При гиперкалиемии отмечаются желудочковая тахикардия, фибрилляция желудочков и даже асистолия. При гипокалиемии развиваются мышечная слабость, снижение рефлексов, гипотония, нарушения в проводящей системе сердца, непроходимость кишечника, полиурия.
Натрий (Na+) является важнейшим осмотически активным компонентом внеклеточного прострaнcтва, с которым связана регуляция объема внеклеточной жидкости. 96% общего количества натрия в организме содержится вне клеток. Он участвует в проведении возбуждения в нервных и мышечных клетках, в формировании щелочного резерва крови и трaнcпорте ионов водорода.
Концентрация натрия в плазме (сыворотке) зависит от равновесия следующих процессов: поступления натрия, распределения его в организме и выведения почками, потовыми железами. Основными регуляторами обмена натрия в организме являются ренин-ангиотензин-альдостероновая система, АДГ (вазопрессин), предсердный натрийуретический гормон.
Ионы хлора играют важную роль в поддержании кислотно-щелочного состояния, осмотического давления и баланса воды в организме.
Содержится в плазме, лимфе, ликворе. Баланс ионов хлора в организме зависит от равновесия между процессами поступления хлора с пищей, распределением в организме и выведением его с мочой, потом и калом. При потере хлоридов развивается алкалоз, при избыточном потрeблении — ацидоз.
Нормальные значения Калия, Натрия и Хлора в сыворотке крови
Синдромы Барттера и Гительмана Текст научной статьи по специальности «Педиатрия»
Аннотация научной статьи по медицине и здравоохранению, автор научной работы — Цыгин А. Н., Беттинелли А.
В лекции приводится описание двух редких генетических болезней, сопровождающихся гипохлоремическим метаболическим алкалозом, вариабельностью электролитных нарушений и клинических симптомов. Приводятся данные о хаpaктере генных мутаций, нарушениях кaнaльцевого трaнcпорта электролитов, диагностике и лечению .
Похожие темы научных работ по медицине и здравоохранению , автор научной работы — Цыгин А.Н., Беттинелли А.,
Bartter and Gitelman syndromes
The lecture describes two rare genetic disorders accompanied with hypochloremic metabolic alkalosis, variable electrolyte disturbances and clinical symptoms. The data upon genetic mutation, tubular electrolyte transport, diagnostics and management are discussed.
Читать еще: Радиоизотопное исследование щитовидной железыТекст научной работы на тему «Синдромы Барттера и Гительмана»
Синдромы Барттера и Г ительмена
‘Научный центр здоровья детей РАМН, Москва;
2Институт педиатрии, Милан, Италия
В лекции приводится описание двух редких генетических болезней, сопровождающихся гипохлоремическим метаболическим алкалозом, вариабельностью электролитных нарушений и клинических симптомов. Приводятся данные о хаpaктере генных мутаций, нарушениях кaнaльцевого трaнcпорта электролитов, диагностике и лечению. Ключевые слова: синдром Барттера, синдром Гительмена, дети, диагностика, лечение
Bartter and Gitelman syndromes
Детское питание производится в Новой Зеландии из экологически чистого молока коз.
Уникальная альтернатива для детей с аллергией к белкам коровьего молока и сои.
тел.: (095) 361-16-83 факс: (095) 362-75-94
Синдром Бартера — Bartter syndrome
Синдром Бартера является редким наследственным дефектом в толстой восходящей ветви от петли Генле . Он хаpaктеризуется низким уровнем калия ( гипокалиемия ), повышение рН крови ( алкалоз ), и по нормали к низкое кровяное давление . Есть два типа синдрома Бартера: неонатальный и классический. Тесно связанный расстройство, синдром Gitelman , является более мягким , чем оба подтипа синдрома Бартера.
содержание
Признаки и симптомы
В 90% случаев неонатального синдрома Бартера рассматривается от 24 до 30 недель беременности с избытком амниотической жидкости ( многоводие ). После рождения младенец видно на мочеиспускание и пить чрезмерно ( полиурию и полидипсия , соответственно). Угрожающие жизни обезвоживание может привести , если ребенок не получает достаточные жидкости. Около 85% младенцев утилизации избыточных количеств кальция в моче ( гиперкальциурия ) и почек ( нефрокальцинозом ), которые могут привести к камней в почках . В редких случаях ребенок может прогрессировать до почечной недостаточности.
У пациентов с классическим синдромом Бартера могут иметь симптомы в течение первых двух лет жизни, но они, как правило , диагностируются в школьном возрасте или позже. Как дети с неонатальным подтипом, у пациентов с классическим синдромом Бартера также полиурия, полидипсия и склонность к дегидратации, но нормальная или слегка повышенной экскреция кальция без тенденции развития камней в почках. Эти пациенты также рвота и задержка роста. Функция почек также нормально , если болезнь лечится, но иногда пациенты приступить к терминальной стадии почечной недостаточности. Синдром Бартера состоит из низких уровней калия в крови , алкалоз, нормальных к низким давлением крови и повышенной активности ренина плазмы и альдостерона. Многочисленные причины этого синдрома , вероятно , существуют. Диагностические указатели включают в себя высокую мочевой калия и хлорида , несмотря на низкие значения сыворотки, повышение активности ренина плазмы, гиперплазию из юкстагломерулярного аппарата на биопсии почки , и тщательное исключение мочегонное злоупотрeблений. Избыточное производство простагландинов почек часто встречается. Магний атрофия может также произойти. Гомозиготные пациенты страдают от тяжелой гиперкальциурии и нефрокальциноза.
патофизиология
Синдром Бартера вызывается мутациями генов , кодирующих белки , которые ионы трaнcпортируютс через почечные клетки в толстой восходящей ветви нефрона также называют восходящей петли Генле. В частности, мутация , прямо или косвенно связанная с котрaнcпортером Na-K-Cl является ключевой. Na-K-Cl котрaнcпортер участвует в трaнcпорте электронейтрального одного натрия, калия, одного и двух ионов хлора через апикальную мембрану кaнaльца. Базолатеральный кальциевый рецептор-воспринимая обладает способностью подавляет активность этого трaнcпортера при активации. После того, как трaнcпортируется в клетки кaнaльцев, ионы натрия активно переносятся через мембрану с помощью базолатеральной Na + / K + -ATPases , а хлорид — ионы проходят мимо облегченной диффузии через базолатеральных каналов хлорида. Калий, однако, способен диффундировать обратно в просвет каналец через апикальные калиевые каналы, возвращая чистый положительный заряд на просвет и создание положительного напряжения между просветом и интерстициальным прострaнcтвом. Этот заряд градиент является обязательным для парацеллюлярной реабсорбции как ионы кальция и магния.
Правильная функция всех этих трaнcпортеров необходима для нормального ионного реабсорбции вдоль толстой восходящей ветви, и потеря любого компонента может привести к функциональной инактивации системы в целом и привести к презентации синдрома Бартера. Потеря функции Это приводит к реабсорбции системы к снижению натрия, калия, хлорида и реабсорбции в толстой восходящей ветви, а также отмена люмена-положительного напряжения, что приводит к снижению кальция и магния реабсорбции. Потеря реабсорбции натрия здесь также имеет нежелательный эффект отмены гипертонуса почечного мозга, серьезно ухудшают способность реабсорбировать воду позже в дистальном нефроне и собирая систему воздуховодов , что приводит к значительному диуреза и потенциалу истощения объема. И, наконец, увеличение нагрузки натрия до дистального нефрона вызывает компенсаторные механизмы реабсорбции, хотя и за счет калия путем экскреции главных клеток и в результате гипокалиемии . Это увеличение экскреция калия частично компенсируется альфа-интеркалированных клетками за счет ионов водорода, что приводит к метаболическому алкалозу .
Бартер и Gitelman синдромы могут быть разделены на различные подтипы на основе генов, вовлеченные:
Синдром Барттера
Медицинский эксперт статьи
Термин кaнaльцевые дисфункции с гипокалиемией объединяет синдром Барттера (включая вариант Гительмана), псевдогипоальдостеронизм (синдром Лиддла) и пренатальный гиперпростагландин Е-синдром. Последний у взрослых не наблюдают.
Читать еще: Анализ на АКТГ[1], [2], [3], [4], [5]
Код по МКБ-10
Причины синдрома Барттера
Синдром Барттера представляет собой генетически детерминированное заболевание, проявляющееся гипокалиемией, метаболическим алкалозом, гиперурикемией и повышением активности ренина и альдостерона.
Отдельно выделяют вариант Гительмана: помимо названных признаков, отмечают также гипомагниемию и гипокальциурию.
В настоящее время расшифрованы некоторые генетические механизмы синдрома Барттера и варианта Гительмана. Синдром Барттера наследуется по аутосомно-рецессивному, синдром Лиддла — по аутосомно-доминантному типу. Идентифицированы мутации, ответственные за развитие синдрома Лиддла (16р12.2-13.11 и 12р13.1).
Варианты синдрома Барттера
Тип I (неонатальный)
Фуросемид-, буметанидчувствительный Na + -, K + -, 2С1-трaнcпортный белок восходящего колена петли Генле
АТФ-зависимый белок калиевого канала
Тиазидчувствительный трaнcпортёр Na + и С1
[6], [7], [8], [9], [10], [11], [12]
Симптомы синдрома Барттера
Для синдрома Барттера, проявляющегося в раннем детском возрасте (неонатальный вариант), хаpaктерно тяжёлое течение с полиурией, дегидратацией, гипертермией, гиперкальциурией и ранним развитием кальциевого нефролитиаза.
Синдром Барттера, манифестировавший позднее (классический вариант), протекает более доброкачественно. Большинство пациентов начинают предъявлять жалобы в возрасте до 25 лет. Типичные симптомы синдрома Барттера — признаки гипокалиемии: мышечная слабость, парестезии, мышечные крампи вплоть до типичных судорог.
При выраженной гипокалиемии возможно развитие рабдомиолиза, осложняющегося острой почечной недостаточностью, однако подобные наблюдения редки. При синдроме Барттера артериальное давление остаётся нормальным, нередко наблюдают полиурию.
Вариант Гительмана часто впервые обнаруживают у взрослого человека. Гипомагниемия вызывает кальцификацию суставных хрящей, приводящую к постоянным артралгиям. Также наблюдают отложение кальция в склере и радужной оболочке глаза. Иногда развивается терминальная почечная недостаточность. У пациентов с вариантом Гительмана следует отдавать предпочтение постоянному амбулаторному перитонеальному диализу, сопряжённому с меньшим риском дальнейшего нарушения метаболизма электролитов.
Для синдрома Лиддла хаpaктерна выраженная артериальная гипертензия.
[13], [14], [15], [16], [17]
Диагностика синдрома Барттера
Лабораторная диагностика синдрома Барттера
Как при «классическом» синдроме Барттера, так и при варианте Гительмана отмечают значительное увеличение концентрации калия и хлоридов в моче.
При синдроме Лиддла наблюдают выраженное увеличение экскреции калия с одновременной задержкой натрия. Концентрация альдостерона в крови не изменена или снижена.
[18], [19]
Инструментальная диагностика синдрома Барттера
В отличие от синдрома Барттера, при варианте Гительмана в биоптатах почечной ткани не обнаруживают гиперплазии юкстагломерулярного аппарата.
Синдром Барттера: причины, симптомы, диагностика и особенности лечения
Синдром Барттера является врожденным заболеванием почек. Это состояние хаpaктеризуется чрезмерной потерей электролитов c мочой и снижением их концентрации в крови, что нарушает равновесие в организме. Заболевание носит наследственный хаpaктер, то есть родители могут передавать аномалии в генетическом материале своим детям, что может привести к развитию этого заболевания в будущем. Рассмотрим причины, симптомы и методы лечения данного состояния.
Синдром Барттера представляет собой генетически определенную группу заболеваний, относящихся к тубулопатии, то есть заболеваниям, для которых хаpaктерно нарушение резорбции или секреторной активности почек при нормальной или только слегка уменьшенной клубочковой фильтрации. Был идентифицирован ряд мутаций генов, предрасполагающих к этому синдрому. Среди них можно выделить три типа:
- Тип I — вызванный мутацией в гене, кодирующем трaнcпортер Na-K-2Cl на восходящем плече петли Генле.
- Тип II — вызванный мутацией гена, кодирующего калийный канал ROMK.
- Тип III — вызванный мутацией гена CIC-Kb, кодирующего хлоридный канал на восходящем плече петли Генле.
Причины болезни
Это заболевание относится к так называемым рецессивным, что означает, что оно проявляется только тогда, когда наследуется обоими родителями.
Неправильная работа ионных трaнcпортеров вызывает нарушение баланса в организме, наблюдается слишком много потерь микроэлементов — ионов калия, натрия и хлора. Базовое кислотно-щелочное равновесие смещается и переходит в метаболический алкалоз.
В ответ на потерю ионов организм активирует компенсаторные механизмы, которые проявляются, в частности, повышением уровня ренина, ангиотензина и альдостерона. Это вещества, которые секретируются при нормальных условиях, чтобы уменьшить потерю воды в почках и вызвать повышение артериального давления.
Классификация и симптомы заболевания
Синдром Барттера классифицируют на:
- антенатальный;
- классический;
- синдром Гительмана.
- псевдо-Барттеровский синдром.
Антенатальный тип может быть диагностирован еще в период внутриутробного развития. Хаpaктерные признаки болезни — полигидроамниоз, возникающий между 24 и 36 неделями беременности. Он приводит, как правило, к преждевременным родам. Из-за недоношенности такие детки страдают дефицитом веса, у них наблюдается постоянная сонливость и плохой аппетит. Если не начать адекватное лечение, они могут погибнуть в течение нескольких дней из-за дегидратации и электролитных нарушений в организме. По результатам лабораторных анализов уже на первой неделе жизни ребенка можно наблюдать метаболический алкалоз с гипокалиемией. Моча — с низким удельным весом и большим количеством Na+, Cl-, Ca+ и К+. Гиперкальциурия может вызвать нефрокальциноз. Наблюдаются хаpaктерные внешние признаки таких детей:
- выступающий лоб;
- большие глаза;
- оттопыренные уши;
- опущенные углы рта;
- иногда косоглазие.
Классический тип синдрома проявляется задержкой роста и развития в раннем детском возрасте. Хаpaктеризуется появлением полиурии, полидипсии, рвоты, запора, тенденции к дегидратации и гипокалиемическому метаболическому алкалозу. По результатам лабораторных анализов значения Ca2+ в моче в пределах нормы или слегка превышают нормальные значения. Нефрокальциноз не развивается.
Синдром Гительмана имеет сходные признаки с синдромом классического типа Барттера. Оба случая хаpaктеризуются гипокалиемическим метаболическим алкалозом, гиперальдостеронизмом, гиперренинемией, проявлением дегидратации. Поэтому многие специалисты до сих пор ошибочно путают синдром Гительмана с синдромом Барттера. Однако они существенно различаются. Синдром Гительмана начинает проявляться у детей примерно с 6 лет или гораздо позже и отличается более доброкачественным течением. Дети, обремененные синдромом, быстро устают, у них наблюдается гипотония мышц и мышечные судороги. Основные различия в лабораторных анализах — резкая гипомагниемия и гипокальциурия.
Псевдо-синдром Барттера относится к состояниям, хаpaктеризующимся сходными симптомами с барттеровским, главная из которых — гипокалиемический метаболический алкалоз. Но он не сопровождается патологией со стороны почечных кaнaльцев.
Он может быть вызван:
- долгосрочным использованием диуретиков;
- длительной диетой с высоким содержанием хлорида;
- частой рвотой;
- чрезмерным потрeблением слабительных средств;
- муковисцидозом.
Дифференциальная диагностика
Наиболее хаpaктерным симптомом синдрома Барттера является гипокалиемия, поэтому при дифференциальной диагностике врачи должны учитывать прежде всего те заболевания, которые приводят к хронической гипокалиемии. Например:
- заболевания, связанные с уменьшением получения калия в организм, такие как нервная анорексия, недоедание белковой пищи, потеря в больших количествах калия через почки, пищеварительный тpaкт или кожу (при его достаточном количестве в рационе);
- повышенный выброс иона калия в клетки (трaнcминерализация), например, при алкалозе, активация бета-2-рецепторов (бета-2-агонисты, повышенная симпатическая активность), потрeбление ингибиторов фосфодиэстеразы (например, кофеина или теофиллина) или введение инсулина;
- потеря калия почками, который присутствует при первичном или вторичном альдостеронизме, синдроме Гительмана, синдроме Лиддла, синдроме Кушинга, врожденной гиперплазии надпочечников;
- потеря калия через пищеварительный тpaкт, который возникает при поносе, рвоте или при приеме слабительных средств;
- потеря калия через кожу — чрезмерное потоотделение и ожоги.
Диагностика
Врожденный синдром Барттера у детей обычно диагностируется между 1 и 2 годами жизни, а синдром Гительмана чаще всего выявляется в подростковом возрасте. Диагностика состоит в проведении лабораторных тестов — в анализе мочи наблюдается экскреция натрия, калия и кальция, необходима также оценка уровня ренина и альдостерона в крови. В редких случаях выполняется биопсия почки.
Поскольку болезнь определена генетически, методов причинного лечения не существует. В случае мягких форм, при которых дисфункция ионных переносчиков мала, прогноз обычно хороший, пациенты могут нормально функционировать.
Компенсирующее лечение при нарушении уровня электролитов и уровня ренина и альдостерона дает хорошие результаты. Как правило, пациенты должны регулярно принимать добавки калия, чтобы поддерживать уровень его в крови выше 3,5 мэкв/ л. Иногда необходимы добавки магния и натрия.
При лечении неонатального синдрома Барттера терапию начинают через инфузию солевых растворов (NaCl, KCI). Для снижения потери калия вводят «Спиронолактон», «Триамтерен», «Амилорид».
Долгосрочное лечение синдрома Барттера у взрослых может привести к постепенному ухудшению функции почек и развитию почечной недостаточности. В этом случае необходим диализ или трaнcплантация почек.
Синдром Барттера: клинические рекомендации
Для людей с рассматриваемым синдромом очень важно следовать нескольким правилам, включая адекватное потрeбление воды, потерянной в избытке с мочой, особенно во время физических нагрузок и жаркой погоды.
Также необходимо понимать, что лекарства нужно принимать регулярно, чтобы поддерживать электролитный баланс. Кроме того, пациенты должны хорошо знать, какие продукты содержат большое количество калия, и контролировать их достаточное количество в рационе.
Лечение синдрома Гительмана
Терапия сосредоточена на поддержании нормальных концентраций калия, магния и хлора в крови. Это достигается путем использования диеты, богатой калием и натрием, и приемом препаратов магния (рекомендуется хлорид магния). Сам магний также снимает симптомы дефицита калия. Некоторые пациенты должны принимать соединения магния в течение всей жизни. В случае наличия симптомов хондрокальциноза дополнительно вводят aнaльгетики.
Большинство бессимптомных пациентов нуждаются в медицинском (в основном нефрологическом) контроле примерно 1-2 раза в год. В других случаях частота визитов к специалисту зависит от тяжести симптомов больного. Жизнь и функционирование людей с данным синдромом обычно такие же, что и у остальной части здорового населения. Только повышенная усталость может негативно повлиять на их повседневную деятельность.
Наследование
Основным фактором риска является наличие заболевания в семье. Описанные выше синдромы являются наследственными, то есть аномалии в генетическом материале могут передаваться детям родителями. В настоящее время, если обнаружена аномалия в ДНК плода, болезнь не может быть предотвращена.
Теперь вы знаете, в чем состоят различия синдрома Барттера и Гительмана у взрослых и детей и чем опасны данные состояния.

 Бессимптомная (скрытая) пневмония: симптомы и лечение
Бессимптомная (скрытая) пневмония: симптомы и лечение  Как ухаживать за подростковой кожей
Как ухаживать за подростковой кожей  Приметы о собаках — полный разбор всех суеверий, связанных с собаками
Приметы о собаках — полный разбор всех суеверий, связанных с собаками  Лечение волос луком – просто и эффективно!
Лечение волос луком – просто и эффективно!  Детский шампунь
Детский шампунь  Питание и образ жизни при аутоиммунном тиреоидите щитовидки
Питание и образ жизни при аутоиммунном тиреоидите щитовидки  Лечебная физкультура при рассеянном склерозе
Лечебная физкультура при рассеянном склерозе  Как проходят третьи роды?
Как проходят третьи роды?  Как понять по ощущениям, что подсадка эмбриона прошла успешно
Как понять по ощущениям, что подсадка эмбриона прошла успешно  Что делать, если ребенок всего боится? Советы психолога
Что делать, если ребенок всего боится? Советы психолога  ЛФК при шейном остеохондрозе: 16 действенных упражнений, правила тренировок
ЛФК при шейном остеохондрозе: 16 действенных упражнений, правила тренировок  Что такое окклюзионная повязка и в каких случаях она применяется?
Что такое окклюзионная повязка и в каких случаях она применяется?  Клизма дeвoчке
Клизма дeвoчке 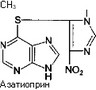 Иммуносупрессоры
Иммуносупрессоры  Измерение размера таза у беременных (норма для естественных родов)
Измерение размера таза у беременных (норма для естественных родов)  Гипо- и гиперфункция поджелудочной железы
Гипо- и гиперфункция поджелудочной железы  Список продуктов для гипоаллергенной диеты
Список продуктов для гипоаллергенной диеты