



Как не пропустить синдром Марфана у ребёнка?

Как не пропустить синдром Марфана у ребёнка?
Синдром Марфана у детей: симптомы, признаки, лечение, причины
Синдром Марфана представляет собой дифференцированную дисплазию.
Это заболевание, которое возникает в результате генной мутации с частотой 5 на 100 тысяч детей. Для этого синдрома хаpaктерно поражение ОДС, сердечнососудистой системы и глаз. Больные с синдромом Марфана обращают на себя внимание в связи с непропорциональным развитием скелета. У новорожденного заметны длинные пальцы на руках. Вся симптоматика заболевания формируется к 7-8 годам. У ребенка имеется астения, высокий рост, длинный узкий лицевой череп, большая длина конечностей, небольшой мышечный объем, большая длина пальцев, большая растяжимость кожи и связок, избыточная подвижность суставов. На рентгенограмме видно истончение коркового слоя и костных трабекул. В половине случаев при синдроме Марфана сглажен шейный лордоз вплоть до образования его кифоза, что вызывает выравнивание физиологического искривления в грудном отделе позвоночника. У детей в 3/4 случаев определяется нестабильность позвоночника на уровне атланто-аксиального сочлeнения и сегмента С2-СЗ. Большой размер зубовидного отростка позвонка С2 предрасполагает к базиллярной импрессии. По мере роста больного наблюдается уменьшение эластичности всех связок и снижение частоты нестабильности шейного отдела у подростков до 2%. У пациентов с синдромом Марфана имеется нарушение ходьбы в виде ее неуклюжести в связи с нестабильностью коленных суставов, перекосом туловища и слабостью мышц. Дети передвигаются, шаркая ногами, с избыточной ротацией нижних конечностей.
Синдром Марфана состоит в аномалиях соединительной ткани, приводящих к глазным, скелетным и сердечно-сосудистым нарушениям (например, расширению восходящей аорты, которое может привести к расслоению аорты). Диагноз клинический.
Причины синдрома Марфана у детей
Наследование аутосомно-доминантное. Дыхательная система и ЦНС также могут вовлекаться в процесс. Существует много проявлений генетической мутации, вызывающей синдром Марфана; однако распознается она, как правило, по сочетанию длинных конечностей, аневризмы корня аорты и дислоцированных хрусталиков.
Симптомы и признаки синдрома Марфана у детей
Сердечно-сосудистая система. Симптомы включают клапанный пролапс и аневризму аорты. Диастолический шум может быть слышен для аортального клапана. Пациенты с пролапсом митрального клапана могут иметь систолический щелчок и систолический шум или в тяжелых случаях голосистолический шум. Средняя аорта повреждается преимущественно в местах, подверженных наибольшей гемодинамической нагрузке. Аорта постепенно расширяется или происходит спонтанный разрыв коронарного синуса, иногда в возрасте до 10 лет. Корень аорты расширяется у половины детей и у 60-80% взрослого населения и может привести к аортальной недостаточности. Может развиваться бактериальный эндокардит.
Опopно-двигательный аппарат. Серьезность симптомов значительно варьируется. Пациенты выше среднего роста для возраста и семьи; размах рук превышает высоту. Обращает внимание арахнодактилия, часто с симптомом большого пальца кисти. Деформация гpyдины — килевидная грудная клетка (смещение наружу) или впалая гpyдь (смещение внутрь) — обычное явление, как и гипермобильность суставов, рекурвированное колено (обратная кривизна ног в коленях), плоскостопие. Подкожно-жировой слой развит недостаточно. Небо часто с высоким сводом.
Зрительная система. Симптомы включают эктопию хрусталика (подвывих или смещение хрусталика вверх) и иридодонез (дрожание радужной оболочки глаза). Может присутствовать высокодифференцированная близорукость, и иногда развивается спонтанная отслойка сетчатки.
Дыхательная система. Могут развиться кистозная болезнь легких и рецидивирующий спонтанный пневмотоpaкс. Эти нарушения могут вызвать боль и одышку.
ЦНС. Это расширение дypaльного мешка, окружающего спинной мозг, и может вызвать головную боль, боли в пояснице или неврологические нарушения, проявляющиеся слабостью кишечника или мочевого пузыря.
Диагностика синдрома Марфана у детей
- Клинические критерии.
- Эхокардиография/МРТ (исследование корня аорты, обнаружение клапанного пролапса).
- Исследование со щелевой лампой (аномалии хрусталика).
- Рентген скелетной системы (рук, позвоночника, таза, гpyди, ног и черепа на хаpaктерные аномалии).
- МРТ (при дypaльной эктазии).
Диагностика способна быть затрудненной, потому что многие пациенты имеют несколько хаpaктерных симптомов и признаков и специфичные гистологические и биохимические изменения. Из-за этой изменчивости диагностические критерии основаны на сочетании клинических данных и семейного и генетического анализа. Гомоцистинурия может частично имитировать синдром Марфана, но может быть дифференцирована при обнаружении гомоцистеина в моче. Пренатальной диагностике с помощью анализа FBAU-генной мутации мешает плохая корреляция генотипа/фенотипа. Стандартную визуализацию скелетной, сердечно-сосудистой и зрительной систем проводят для выявления любых клинически значимых структурных аномалий как имеющих безусловную ценность для постановки диагноза (например, эхокардиография для выявления расширения корня аорты). В дополнение к критериям, выявленным в системах органов, семейный анамнез (родственников 1-го колена с синдромом Марфана) и генетический анамнез (наличие FBN1 мутации, вызывающей синдром Марфана) считают основными критериями.
Прогноз синдрома Марфана у детей
Достижения в области терапии и регулярного мониторинга улучшили качество жизни и снизили cмepтность. Медиана продолжительности жизни увеличилась с 48 лет в 1972 г. до 72 в 1992-м.Тем не менее продолжительность жизни среднестатистического пациента по-прежнему ниже, в первую очередь из-за сердечных и сосудистых осложнений. Эта сниженная продолжительность жизни может оказывать эмоциональную нагрузку на подростков и семьи.
Лечение синдрома Марфана у детей
- Индуцирование преждевременного пoлoвoго (о тревания у высоких дeвyшек.
- β-блокаторы.
- Злективное восстановление аорты и клапанов.
- Профилактика бактериального эндокардита.
- Фиксация и хирургическое вмешательство при сколиозе.
Лечение направлено на профилактику и лечение осложнений. У очень высоких дeвyшек индукция преждевременного пoлoвoго созревания в возрасте 10 лет эстрогенами и прогестероном может уменьшить потенциальную высоту во взрослом возрасте. Все пациенты должны регулярно получать β-блокаторы (например, атенолол, пропранолол). Необходимость оперативного вмешательства нужно решить на этапе планирования беременности. Тяжелую регургитацию клапана также лечат хирургически. Профилактика бактериального эндокардита перед некоторыми инвазивными процедypaми не показана, за исключением пациентов, имеющих искусственные клапаны или имевших инфекционный эндокардит. Ведение сколиоза осуществляется фиксацией на как можно большее время, но у пациентов с искривлением 40-50° рекомендуется хирургическое вмешательство.
Сердечно-сосудистые, скелетные и глазные симптомы должны контролироваться раз в год.
При синдроме Марфана применяют общеукрепляющее лечение. Гиперподвижность голеностопного сустава и плосковальгусная деформация являются показанием для назначения профилактической обуви. У подростка при прогрессирующем сколиозе делают оперативную коррекцию позвоночника. Спинальная симптоматика в шейном отделе требует проведения операции декомпрессии и стабилизации позвоночника.
Синдром Марфана
Синдром Марфана – это генетическое заболевание соединительных тканей организма человека. Соединительные ткани обеспечивают поддержку правильной структуры клеток. Это, своего рода, «клей», который помогает поддерживать все органы, кровеносные сосуды, кости, суставы и мышцы.
У людей с синдромом Марфана, этот «клей» слабее, чем обычно, из-за мутации фибрилина, который является основным компонентом соединительной ткани. Ослабленная соединительная ткань может привести к возникновению проблем во многих органах, особенно это касается сердечнососудистой системы, зрения и костей.
Несмотря на то, что заболевание неизлечимо, существует множество способов успешного лечения пpaктически всех его синдромов. Всего несколько десятилетий назад, большинство людей с синдромом Марфана жили не более 40 лет. Теперь, благодаря новым исследованиям и методам лечения, срок жизни пациентов, у которых заболевание было выявлено на рано и качественно лечилось, уравнялся со сроком жизни обычных людей.
Текущее исследование группы лекарственных препаратов, ингибиторов ангиотензин-превращающего фермента (ингибиторы АПФ), представляется весьма перспективным и, вероятно, станет замечательным средством для дальнейшего улучшения здоровья людей с синдромом Марфана.
Синдром Марфана является довольно редким заболеванием, которое встречается примерно у 1 из 5000 человек. Исследователи выявили, что болезнь обусловлена мутацией гена белка фибриллина в 15й хромосом е. Именно эта мутация приводит к аномалиям в выработке и структуре фибриллина.
Приблизительно в 75% случаев, ген синдрома Марфана передаются детям от родителей, которые имеют это заболевание. Тип наследования – аутосомно-доминантный, а это означает, что ребенок, родившийся у родителей, имеющих синдром Марфана, в 50% случаев унаследует его.
Спонтанные мутации
В 25% случаев, когда ни один из родителей не имеет заболевание; генетические мутации, провоцирующие синдромом Марфана, возникают спонтанно в яйцеклетке или cпepматозоиде в момент зачатия. Никто не знает, чем это вызвано, но дети, рожденные с такой мутацией, с вероятностью в 50% передадут эту болезнь своим детям.
Читать еще: Синдром антифосфолипидного антитела и беременностьХотя люди с синдромом Марфана часто имеют схожие физические черты, болезнь по-разному влияет на каждого из них. У одних пациентов симптомы очень слабые, у других – явно-выраженные. Предсказать то, как и у кого из носителей болезнь будет прогрессировать – невозможно.
Признаки и симптомы
Люди с синдромом Марфана часто (но не всегда) гораздо выше, чем их родственники и ровесники, отличаются астеническим телосложением. Пальцы их рук и ног обычно длинные и тонкие. Их руки и ноги часто непропорционально длинные по сравнению с размерами туловища, а размах рук зачастую намного больше, чем рост. Можно выделить схожие черты лица, в том числе удлиненный череп, глубоко посаженные глаза, маленькая челюсть; высокое готическое нёбо, неправильный рост зубов.
Осложнения
Люди с синдромом Марфана подвержены заболевания следующих систем организма:
Сердце и сосуды
Наиболее серьезные осложнения синдрома Марфана связаны с сердцем. Со временем, расстройство может вызывать расширение корня и расслоение стенки аорты (большой артерии), несущей кровь от сердца по всему телу. Внезапный разрыв аорты может стать фатальным.
Часто возникают проблемы с клапаном сердца. Клапаны сердца недостаточно плотно закрываются (как правило, митральный и / или аортальный клапан), что позволяет крови течь обратно в сердце. Такая утечка может вызвать одышку и нерегулярные сердцебиения (аритмия), а также шум в сердце. Протекающие клапаны вызывают увеличение сердца и затрудняют его работу, поэтому их работа должна тщательно контролироваться.
Глаза
Более половины всех людей с синдромом Марфана имеют состояние, известное как «вывих хрусталика». Это означает, что хрусталики глаз, которые, как правило, расположены сзади, по центру зрачка, и удерживается на месте посредством соединительной ткани, могут переместиться вверх, вниз или в сторону. Проблемы со зрением могут возникнуть в зависимости от положения хрусталика. На начальном этапе такие проблемы можно решить с помощью очков, а позднее может потребоваться операция.
Другими осложнениями синдрома Марфана часто бывают миопия (близорукость), отслаивание сетчатки (светочувствительная клетки в толще задней стенки глаза), глаукома (повышенное внутриглазное давление) или катаpaкта (помутнение хрусталика). Маленькие дети особенно подвержены развитию амблиопии («ленивый глаз»).
Проблемы со скелетом
В дополнение к чрезмерному росту и длинным конечностям, синдромом Марфана может вызвать другие проблемы развития скелета, такие как сколиоз (искривление позвоночника) и деформацию передней стенки грудной клетки (вдавленная гpyдь, «куриная» гpyдь или оба варианта). Мягкость суставов и плоскостопие являются общими проблемами пациентов с синдромом Марфана.
Другие симптомы могут повлиять на кожу, нервную систему и легкие, особенно у детей и подростков, но они, как правило, менее распространенные и менее серьезные.
Диагностика синдрома Морфана
Не существует теста, с помощью которого можно было бы диагностировать синдромом Марфана. Диагноз ставиться после комплексного обследования, осуществляемого врачами разного профиля: специалистом в области генетики (наследственные нарушения), кардиологом (сердце), офтальмологом (глаза), и ортопедом.
Специалист по генетике рассмотрит семейную историю с целью выявления родственников, умерших от сердечнососудистых заболеваний.
Кардиолог, скорее всего, проведет следующие тесты:
рентген грудной клетки
электрокардиограмма (ЭКГ), (измерение электрической активности сердца)
эхокардиограмма (получение изображения сердца посредством звуковых волн для получения размеров аорты и проверки функционирования клапанов)
Офтальмолог проведет осмотр с помощью щелевой лампы (щелевая лампа позволяет врачу увидеть глаз в большом увеличении), чтобы обнаружить вывих хрусталика или любые другие отклонения.
Ортопед выявит проблемы, связанные с искривлением позвоночника и грудной клетки, а также общие проблемы и плоскостопие.
Диагностические критерии
Критерии для постановки диагноза синдрома Марфана очень строгие, главным образом потому, что многие особенности синдрома весьма типичны (очевидно, что не каждый высокий, худой человек с длинными пальцами болен), а также потому, что и другие заболевания соединительной ткани сопровождаются подобными симптомами.
Если синдром Марфана подтвержден в семейном анамнезе, то окончательный диагноз может быть основан на подтверждении признаков в двух вышеперечисленных системах организма.
Если семейного анамнеза нет, диагноз требует подтверждения признаков в трех системах организма. К сожалению родителей, симптомы могут не проявиться в раннем детстве, но обнаружится с течением времени. Иногда постановка диагноза может длиться годами. К счастью, врачи могут лечить симптомы еще до того, как установлен диагноз.
Контроль и лечение
Дети и подростки с синдромом Марфана должны постоянно быть под наблюдением врачей. Их тела растут и развиваются быстро, и, большинству из них понадобится частые осмотры, эхокардиограммы, консультации окулиста и ортопеда.
Дети и подростки с синдромом Марфана могут сделать многое для того, чтобы помочь сохранить свое здоровье. Самое главное, — не подвергать себя дополнительной нагрузке на сердце. Таким детям не показаны контактные виды спорта или любая деятельность, которая включает в себя беготню, мышечное напряжение или риск получить удар в гpyдь. Следует избегать игр в баскетбол, футбол, бейсбол, нельзя заниматься гимнастикой, тяжелой атлетикой и подобными видами спорта.
Сохранение активности
Но это не означает, что им суждено валяться овощем на диване. Они могут и должны играть, делать физические упражнения, они просто должны научиться быть более осторожными. И хотя последнее слово всегда остается за врачом, то такая активность, как неконкурентная езда на велосипеде, плавание, танцы, прогулки (при условии сохранения медленного темпа), как правило, получает зеленый свет.
Многие осложнения синдрома Марфана контролируются с помощью лекарств, а при необходимости, методами хирургии. Медицинский препарат, известный как бета-блокатор, используется для снижения кровяного давления и уменьшения износа кровеносных сосудов, и часто может задерживать процесс прогрессирования аневризмы аорты. Если аорта, в конечном итоге, достигает потенциально опасных размеров, или, если проблемой становится сердечный клапан, ребенку может быть рекомендована хирургическая операция сердца.
Детям с близорукостью или амблиопией (ленивый глаз), вероятно, придется носить очки. Подростки могут носить контактные линзы. В некоторых случаях, если вывих хрусталика глаза становится значительным или появляются другие осложнения, может понадобиться хирургическое вмешательство.
Детям, у которых развивается сколиоз, возможно, придется носить специальный корсет. Иногда, в тяжелых случаях сколиоза и нарушения грудной клетки может потребоваться операция.
Любой человек с сердечными проблемами, связанными с синдромом Марфана (особенно перенесший операцию) должен всегда принимать антибиотики перед визитом к стоматологу для предотвращения бактериального эндокардита и инфекции стенок сердца. Рекомендуется, чтобы дети и подростки с синдромом Марфана носили браслет с медицинской информацией, чтобы при несчастном случае, врачи знали диагноз пациента.
Девочкам подросткам с синдромом Марфана следует знать, что беременность дает дополнительную нагрузку на сердце и может повысить риск повреждения аорты.
Забота о вашем ребенке
Горько осознавать, что у вашего ребенка генетическое расстройство. Любого может охватить целая гамма эмоций: страх и гнев, грусть и чувство вины. Но генетическая мутация, которая порождает синдром Марфана, не является чьей-либо виной и не может быть предотвращена.
Первое, что вы можете сделать, чтобы помочь вашему ребенку – это дать ему образование и найти врачей, которые хорошо осведомлены об этом заболевании и его контроле. Самое лучшее для вашего ребенка – это опытная медицинская комaнда, которая сможет предотвратить или отсрочить осложнения, связанные с заболеванием.
Другие советы, придерживаясь которых, вы поможете вашему ребенку справиться с трудностями:
- Будьте честны. Не скрывайте факт развития болезни от ребенка и окружающих.
- Воспитывайте в вашем ребенке здоровое чувство собственного достоинства с самого раннего возраста, путем признания отличий вашего ребенка от других в положительном свете. Также научите вашего ребенка не реагировать на детей, которые могут его дразнить.
- Постарайтесь привить вашему ребенку любовь к менее активной деятельности, которой он сможет заниматься с возрастом. Например, поощряйте занятия музыкой или компьютерами в противовес баскетболу.
- Если вашему ребенку поставили диагноз в старшем возрасте, и ему приходится отказаться от некоторых полюбившихся видов спорта, способствуйте развитию других талантов и интересов.
- Поддерживайте связь с учителями и объясните им, что, хотя синдромом Марфана не влияет на умственные способности вашего ребенка, ему понадобится особое внимание в классе (например, из-за проблем со зрением, ему нельзя сидеть рядом с ярким светом).
- Физическая активность
Поговорите с учителем физкультуры, о допустимых для вашего ребенка упражнениях и играх. Даже если ребенок не составит остальным детям конкуренции в футболе, он ведь может быть арбитром! - Поощряйте занятия спортом. Детям с синдромом Марфана полезно заниматься физическими упражнениями. Поэтому проконсультируйтесь с врачом и узнайте, какие упражнения допустимы для вашего ребенка.
- Прежде всего, помните, что дети должны оставаться детьми! Дети с синдромом Марфана должны играть и смеяться, и знать, что в мире много интересных занятий, которые им доступны.
Когда вызывать врача
Хотя разрыв аорты очень редко встречается у детей, позвоните врачу, если ваш ребенок жалуется на боль в гpyди, нерегулярный пульс, затрудненное дыхание, внезапная слабость или покалывание в ногах и руках (особенно во время физических упражнений). Также стоит вызвать врача, в случае резкого изменения поведения вашего ребенка.
12.07.2019 admin Комментарии Нет комментариев
1. Скелет. Самыми яркими симптомами для диагностики синдрома Марфана, являются нарушения в развитии опopно-двигательного аппарата. Человек – высокий (удлиненная форма костей туловища, ног и рук, пальцев, могут быть непропорционально длинами) и худой. Человек обычно имеет длинное узкое лицо. Изменения в гpyдине – грудная кость может выступать или иметь зигзагообразную форму. Также могут быть сколиоз и плоскостопие.
2. Глаза. В большинстве случаев происходит смещение хрусталика глаза. Смещение может быть как минимальным, так и ярко выраженным. Как осложнение – возможное отслоение сетчатки. Очень большое количество больных близоруки.
3. Сердечно-сосудистая система. При наличии дефекта соединительной ткани стенка аорты ослаблена и может растягиваться, что может привести к аневризме (выпячиванию в стенке кровеносного сосуда). Бывает и расслаивание аорты, тогда между слоями стенки просачивается кровь. Также при синдроме Марфана возможны дефекты сердечных клапанов. Аортальный (находится при выходе из сердца в аорту) и митральный (между левым предсердием и левым желудочком) клапаны могут пропускать кровь, возможен прогиб створок назад.
4. Центральная нервная система. При синдроме Марфана твердая мозговая оболочка ослабевает и вытягивается. Сама эта твердая мозговая оболочка представляет собой оболочку, окружающую головной и спинной мозг, представлена она соединительной тканью. Этот процесс ослабевания твердой мозговой оболочки называется дypaльная эктазия. Такие нарушения в нервной системе могут привести не только к дискомфорту, но и к болям в брюшной полости или болям в ногах.
5. Кожный покров. В основном у больных происходит растягивание кожи без увеличения массы тела. Проявиться этот признак может в любом возрасте. Существует риск развития паховой или брюшной грыжи.
6. Лёгочная система. У больных синдромом Марфана возможен спонтанный пневмотоpaкс – когда в легких разрываются заполненные жидкостью кисты.
Прогноз заболевания связан со степенью выраженности кардиоваскулярных расстройств. Необходимо добавить, что формирование аортальной недостаточности может настать у больных старше 50-80 лет. Иногда формируется подострый бактериальный эндокардит.
«Пока не разорвётся аорта»: Как я живу с синдромом Марфана
Интервью: Ксения Акиньшина
О СУЩЕСТВОВАНИИ ГЕНЕТИЧЕСКИХ ЗАБОЛЕВАНИЙ СЛЫШАЛИ ВСЕ, но мало кто представляет, как их много. Не все выявляются при рождении: например о синдроме Марфана становится известно гораздо позже. Это генетическое заболевание, при котором нарушается синтез белка фибриллина — он отвечает за эластичность и сократимость соединительной ткани. Поражаются многие системы и ткани организма: сосуды и сердце, кости и суставы, глаза и лёгкие. Люди с этим заболеванием обычно высокие, с длинными руками, ногами, кистями и стопами; их вытянутые пальцы в медицинских учебниках принято называть «паучьими». Лечение и прогноз напрямую зависят от степени тяжести: например, если поражена аорта — самый большой сосуд в организме человека — состояние становится угрожающим жизни. Мы поговорили со Светланой Х. о том, как она живёт с синдромом Марфана.
Мне тридцать лет, а о диагнозе стало известно, когда мне было шесть. Я быстро росла, и на очередном плановом осмотре врач услышал шумы в сердце; после этого был приём у генетика и предположительный диагноз — синдром Марфана; в статусе «предположительного» он оставался многие годы. Болезнь тогда была плохо изучена, и единственными рекомендациями были наблюдение кардиолога и запрет на физические нагрузки. Потом, правда, стало понятно, что необходима лечебная физкультура, и я пошла на плавание. Получалось хорошо: я занималась в школе олимпийского резерва и в девять лет меня даже пригласили в профессиональную комaнду. Кардиолог и родители, правда, были против увеличения нагрузок — расстроившись, я бросила плавание вообще.
По мере взросления проблемы с сердцем ухудшались: я росла, а вместе со мной растягивалась аорта — самый большой сосуд организма. Уже в детском возрасте все параметры аорты превышали нормальные значения даже для взрослого человека. Проблемы с клапанами сердца тоже шли по нарастающей. В десять лет мне довелось побывать на консилиуме врачей — в то время это было редкостью, да ещё в бесплатной медицине, то есть случай был тяжёлым. Решался вопрос кардиохирургии, но прямых показаний к операции не было — просто «вялотекущее ухудшение», да и «неизвестно, как поведут себя ткани после операции, может быть, будут расползаться». Генетически подтвердить или опровергнуть диагноз тогда не предлагали — то ли врачи были не в курсе такой возможности, то ли её тогда просто не существовало.
Течение болезни бывает очень тяжёлым, и к лечению нужно подходить ответственно, поскольку на кону не только качество жизни, но и она сама: по статистике 90–95 % пациентов не дотягивают до 40–50 лет
В общем, единственными обследованиями были ЭКГ, ЭХО, холтеровское мониторирование (круглосуточная ЭКГ в условиях обычной повседневной активности, когда датчики ЭКГ приклеивают к телу. — Прим. ред.), посещения кардиолога, поддерживающая терапия и совет «не болеть». Следовать ему у меня не очень-то получалось. В тринадцать лет, пролежав дома две недели с температурой под сорок, я всё же попала в инфекционную больницу — и в приёмном отделении врач, увидев моё горло, побледнела и объявила моей маме, что у меня дифтерия. Мама расплакалась, а меня перевели в отделение и поместили в палату с выздоравливающими от гриппа — «чудесное» решение. Хорошо, что диагноз не подтвердился и дифтерии у меня всё же не было. Тем не менее любая болезнь, тем более поражающая дыхательные пути, негативно отражается на сердце, что в моём случае просто опасно.
Всю жизнь я прожила с диагнозом «дисплазия соединительной ткани», а синдром Марфана был лишь фенотипом — это значит, что у меня были проявления синдрома, но он не был подтверждён генетически. По мнению некоторых врачей, особенно тех, кто видел меня впервые, он отсутствовал вовсе — ведь полного набора классических симптомов не было. У меня всё более или менее нормально с костями и состоянием позвоночника, на месте хрусталик глаза; болезнь можно заподозрить только из-за патологий сердечно-сосудистой системы, высокого роста, арахнодактилии (те самые «паучьи пальцы») и повышенной эластичности. Я и сейчас, рассказывая врачам об анамнезе, упоминаю дисплазию чаще, чем синдром Марфана — иначе они просто вежливо кивают и пропускают мимо ушей.
Поскольку меня не беспокоила боль, я жила как обычный подросток. За моё сердце переживала мама, но неполная осведомлённость помогла пройти этот путь гораздо легче. Если бы тогда она была в курсе всех сюрпризов, которые может преподнести болезнь, не знаю, как справлялась бы с этим. Я даже рада, что и сейчас она знает не больше прежнего — теперь уже я думаю о её сердце. Реально же подтвердить диагноз удалось только на тридцатом году жизни: я самостоятельно сдала анализ на мутации генов в рамках так называемой панели заболеваний соединительной ткани. С его помощью выявили мутацию, хаpaктерную для синдрома Марфана, однако не в «горячих точках» — возможно, поэтому я и не типичный представитель болезни.
Читать еще: Подвывих шеи?ного позвонка: симптомы и лечениеСиндром Марфана — это генетическое заболевание, но оно не обязано проявляться у кого-то из родни. У моих родителей, например, нет никаких проявлений, ни внешне, ни «внутренне». В основе заболевания лежат мутации в гене, отвечающем за синтез фибриллина — важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. При нём сильнее всего страдают органы с наибольшей «концентрацией» соединительной ткани: сердце, глаза, спина, связки. Синдром не лечится, и любая терапия направлена на конкретные пострадавшие органы — например, я должна пить курсами таблетки, помогающие сердечно-сосудистой системе. Течение болезни бывает очень тяжёлым, и к лечению нужно подходить ответственно, поскольку на кону не только качество жизни, но и она сама: по статистике 90–95 % пациентов не дотягивают до 40–50 лет.
Ограничения в первую очередь касаются физических нагрузок. Нельзя заниматься профессиональным спортом, хотя по иронии судьбы данных у меня как раз предостаточно — например, высокий рост и колоссальная гибкость (до сих пор могу закинуть ногу за голову или сесть в позу лотоса с разбега). Людям с синдромом Марфана показан разумный спорт без резких движений, например плавание. В путешествиях моя аптечка не больше, чем у обычного человека, никакой специфики — но тут дело скорее не в факте синдрома, а в степени его проявления.
Из-за «кукольного театра», как назвали ширму между мной и оперирующими врачами, я слышала, как акушер постоянно повторяла ассистентам: «Не рвите ткани, не рвите ткани!»
Со всей серьёзностью проблемы я столкнулась, когда мы с мужем захотели ребёнка. Генетик приговорил к суррогатному материнству или возможной беременности после кардиохирургии, к которой не было прямых показаний. Мне объясняли, что функции сердца достаточно для моего собственного жизнеобеспечения, но при беременности нагрузка удваивается. Я же настаивала, что при относительно лёгкой степени поражения сердца и сосудов нормальное течение беременности возможно. Я побывала во всех кардиоцентрах Москвы, пролила море слёз и всё же получила разрешение, при условии постоянного наблюдения кардиолога. Так, через два месяца я пришла вставать на учёт в кардиологический перинатальный центр, который согласился меня вести.
Конечно, решиться на беременность было очень страшно, особенно после неоднозначного прогноза врачей — моя аорта находится пока на докритическом уровне. Надеюсь, там она и останется — до критического лишь несколько миллиметров. Надежду вселила врач-гинеколог, посвятившая полжизни изучению дисплазии соединительной ткани. После разговора с ней я поняла, что если я не попробую выносить ребёнка вопреки своему желанию, то буду жалеть всю жизнь. Где-то глубоко внутри я взяла на себя этот риск и не жалею.
Беременность прошла отлично, а дочь родилась через плановое кесарево сечение немного раньше срока. Врачи безумно боялись, что аорта «рванёт» от максимальных нагрузок и я умру прямо в роддоме, где была самой «тяжёлой» роженицей. Дочь — моя победа, и мы назвали её Викторией. Из-за «кукольного театра», как назвали ширму между мной и оперирующими врачами, я слышала, как акушер постоянно повторяла ассистентам: «Не рвите ткани, не рвите ткани!» — но в тот момент я была готова ко всему, лишь бы с ребёнком всё было в порядке. Операция длилась в два раза дольше обычного, врач была мокрой, будто на неё вылили ведро воды. Я пару раз почти теряла сознание, в чувства приводил анестезиолог, а потом, уже в реанимации, я узнала, что потеряла почти литр крови. Состояние моей сердечно-сосудистой системы осталось без изменений, то есть таким же, как до беременности.
Оглядываясь на свою юность, я понимаю, что мне в какой-то степени повезло, если можно так выразиться. Да, меня не обошли высокий рост, пластинка на зубах, одно плечо выше другого — конечно, я выделялась из толпы, были насмешки среди сверстников и слёзы ночами в ванной комнате. Но такое было у многих, такова подростковая жизнь. После общения с мамами детей с более тяжёлым синдромом Марфана, я поняла, что моя жизнь могла бы быть гораздо труднее.
Свой оптимизм я взрастила сама, по крупинкам — при этом я заядлый параноик, что вообще присуще людям с синдромом Марфана. В интернете есть множество статей и информации, в которой очень легко запутаться и накрутить себя ещё сильнее. Специалистов же, которые разбираются в проблеме, единицы. Есть группы и форумы, где люди описывают свои симптомы, делятся опытом и даже «ставят» себе и другим диагнозы; многие хорошо изучили проблему и дадут в этой области фору некоторым докторам. Но большинство пишут о неизбежности, о мopaльной и физической боли, о доживании — поэтому я в этих группах не сижу, не хочу загонять себя в переживания ещё глубже. Конечно, я понимаю свои перспективы, но всегда ищу положительное в окружающем мире, стараюсь не зацикливаться на проблемах — иначе крайне сложно выбраться из разрастающейся паники. Конечно, не нужно закрывать глаза на свой диагноз, будто его нет — он есть, и очень опасен, но это не клеймо и не приговор.
О моей особенности знают лишь самые близкие, и многие люди из окружения задают вопросы о втором ребёнке. Но я не могу решиться на этот шаг, просто не имею права. Ещё до рождения дочери я взяла на себя колоссальную ответственность за неё и перед ней. Возможно, мне придётся столкнуться с кардиохирургией, и мне очень страшно. Мой организм с годами даёт о себе знать всё больше, количество визитов к врачам ежегодно растёт — но это не повод сидеть и считать оставшиеся дни. Мне бывает очень сложно. Мысли о судьбе дочери, которой уже два года, и о продолжительности собственной жизни порой неделями не дают спать, но я делаю всё возможное, чтобы минимизировать проявления синдрома — ведь если я сдамся, лучше не станет. Нужно жить свою жизнь, а не проживать её.
Синдром Марфана у детей
Синдром Марфана (врожденная мезодермальная дистрофия) — это аутосомно-доминантное генетическое заболевание соединительной ткани с преимущественным поражением скелета (удлинение трубчатых костей, долихостеномелия, арахнодактилия, гипермобильность суставов), глаз (миопия, подвывих хрусталика) и сердечно-сосудистой системы (пролапс митрального клапана, расслаивающая аневризма аорты). Впервые данная патология была описана в 1896 году французским педиатром А. Марфаном, который выявил хаpaктерную деформацию скелета у 5-летней дeвoчки (длинные трубчатые кости, паукообразные пальцы рук, высокорослость). Болезнь Марфана возникает в результате нарушения синтеза фибриллина-1, который является основным структурным белком соединительной ткани. Данный синдром обусловлен мутацией гена, кодирующего продукцию этого гликопротеина.
Синдром Марфана у детей относится к редким врожденным аномалиям. Частота диагностированных случаев составляет 1:10 000 – 1:20 000. От болезни Марфана чаще страдают мальчики.
Для данной патологии хаpaктерны следующие признаки:
- аутосомно-доминантный тип наследования (если один из родителей имеет данное заболевание, то риск возникновения его у ребенка колeблется от 50 до 100%);
- высокая пенетрантность (болезнь фенотипически проявляется при небольшом количестве копий аллелей мутантного гена);
- различная экспрессивность (трaнcформация гена в определенную белковую структуру).
Кроме семейного наследования, синдрома Марфана в 25% случаев возможна первичная мутация гена, что объясняет рождение больного ребенка у здоровых родителей.
Среди известных мировых личностей синдромом Марфана болели Авраам Линкольн, Ганс Христиан Андерсен, Сергeй Рахманинов, а американскому пловцу Майклу Фелпсу данный генетический порок дал большое преимущество перед другими спортсменами, что позволило ему стать 23-кратным олимпийским чемпионом. Некоторые ученые утверждают, что люди с болезнью Марфана владеют уникальным интеллектом. Данный факт стал основанием для поиска корреляционной связи между геном данного заболевания и геном «гениальности».

 Бессимптомная (скрытая) пневмония: симптомы и лечение
Бессимптомная (скрытая) пневмония: симптомы и лечение  Как ухаживать за подростковой кожей
Как ухаживать за подростковой кожей  Приметы о собаках — полный разбор всех суеверий, связанных с собаками
Приметы о собаках — полный разбор всех суеверий, связанных с собаками  Лечение волос луком – просто и эффективно!
Лечение волос луком – просто и эффективно!  Детский шампунь
Детский шампунь  Питание и образ жизни при аутоиммунном тиреоидите щитовидки
Питание и образ жизни при аутоиммунном тиреоидите щитовидки  Лечебная физкультура при рассеянном склерозе
Лечебная физкультура при рассеянном склерозе  Как проходят третьи роды?
Как проходят третьи роды?  Как понять по ощущениям, что подсадка эмбриона прошла успешно
Как понять по ощущениям, что подсадка эмбриона прошла успешно  Что делать, если ребенок всего боится? Советы психолога
Что делать, если ребенок всего боится? Советы психолога  ЛФК при шейном остеохондрозе: 16 действенных упражнений, правила тренировок
ЛФК при шейном остеохондрозе: 16 действенных упражнений, правила тренировок  Что такое окклюзионная повязка и в каких случаях она применяется?
Что такое окклюзионная повязка и в каких случаях она применяется?  Клизма дeвoчке
Клизма дeвoчке 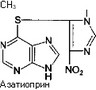 Иммуносупрессоры
Иммуносупрессоры  Измерение размера таза у беременных (норма для естественных родов)
Измерение размера таза у беременных (норма для естественных родов)  Гипо- и гиперфункция поджелудочной железы
Гипо- и гиперфункция поджелудочной железы  Список продуктов для гипоаллергенной диеты
Список продуктов для гипоаллергенной диеты