



Синдром Стерджа-Вебера

Синдром Стерджа-Вебера
Синдром Стерджа — Вебера — Краббе: симптомы, причины, диагностика и лечение
- 26 Июня, 2018
- Неврология
- Яна Троянова
В 21-м веке значительно возрос процент заболеваний неврологическими недугами, которые ранее либо не существовали, либо очень редко давали о себе знать. К последней категории относится опасный для жизни человека синдром Стерджа — Вебера, хаpaктеризующийся определенными особенностями, с которыми необходимо ознакомиться каждому, кто столкнулся с данным заболеванием и желает узнать своего врага в лицо.
Краткая хаpaктеристика недуга
Синдром Стерджа — Вебера — Краббе (иначе: энцефалотригеминальный ангиоматоз) относится к редкой системной патологии неврологического хаpaктера. Тяжелый недуг нередко сопровождается появлением опухолевидных элементов на лицевой части, а также поражением мягкой оболочки головного мозга и глазных яблок. В качестве спутников опасной болезни выступают серьезные изменения, которые могут способствовать проявлению необратимых процессов в организме человека.
Впервые рассматриваемый синдром был выявлен в далеком 1879 году. Несмотря на относительную отдаленность печального события, до сего момента болезнь считается малоизученной: медицинские сведения о ней достаточно скудны. Анамнез заболевания неутешителен.
Распространенность
Согласно статистике, синдром Стерджа — Вебера регистрируется нечасто. Среди 85-100 тысяч новорожденных встречается, как правило, 1 человек, у которого проявляются соответствующие симпомы.
Если у малыша наблюдается пламенеющий невус (или винные пятна) в области глаз, вероятность его заболевания значительно повышается. В 7-8 % подобных случаев ребенок наследует опасный недуг.
Как мужчины, так и женщины подвержены заболеванию в равном процентном соотношении. Будьте внимательны.
Причины проявления
Стоит отметить, что представители медицинской отрасли, изучающие синдром Стерджа — Вебера, разделились на два лагеря. В каждом из них существует диаметрально противоположная точка зрения относительно основной причины проявления болезни. Одни считают, что синдром способен передаваться по наследству (наиболее распространенная версия), а другие придерживаются иного мнения.
Считается, что заболевание связано с неправильным развитием плода на эмбриональной стадии. В момент сбоя нарушается формирование 2 зародышевых лепестков: эктодермы и мезодермы, отвечающих за формирование внутренних органов, кожных покровов, различных сосудов и нервной системы.
Врачи-специалисты утверждают, что проявление недуга напрямую связано с отрицательным воздействием на будущего малыша определенных факторов. К таковым относятся:
- распитие алкогольных напитков;
- употрeбление наркотических веществ;
- курение (данный пункт с высокой долей вероятности заявит о себе, если вредная привычка имела место на ранних сроках беременности);
- наличие инфекций, передающихся пoлoвым путем;
- серьезный сбой в обмене веществ будущей матери, связанный с хроническим недостатком гормонов щитовидки;
- употрeбление медикаментозных препаратов без необходимого контроля;
- возникновение внутриутробных инфекций.
Некоторые люди выражают беспокойство, задумавшись о возможном способе передачи синдрома, однако не стоит переживать. На данный момент не существует официального подтверждения заражения болезнью в обычной жизни (воздушно-капельным путем или при непосредственном контакте с заболевшим).
Возможные симптомы
Недуг хаpaктеризуется неутешительными признаками, которые способны негативно влиять на работу организма человека. Помимо прочих симптомов синдрома Стерджа — Вебера выделяют классическую триаду:
- глаукома врожденного типа, проявляющаяся в нарушении зрительной функции по причине внутриглазного давления;
- сосудистые опухоли в области лица — ангиомы (хорошо диагностируются при УЗИ глаза);
- опухолевидные новообразования сосудистой оболочки головного мозга.
Также в анамнезе заболевания могут наблюдаться:
- нарушение произвольной двигательной функции (парез);
- синдром дефицита внимания и гипеpaктивности;
- умственная отсталость;
- эпилепсия, выраженная неожиданными судорожными приступами;
- гемианопсия, связанная с наличием патологии в области пересечения волокнистых элементов зрительных нервов;
- разрастающийся на лице невус;
- сильные головные боли;
- гидроцефалия или водянка головного мозга;
- олигофрения или идиотия;
- выраженная слабость в мышцах рук и ног;
- низкая чувствительность в непосредственной близости от пораженных участков тела;
- эмоциональный дисбаланс;
- увеличение глазных яблок;
- слепота;
- деформация одной половины лица (гемиатрофия).
В подавляющем большинстве случаев у заболевших людей могут проявиться серьезные поражения слизистых оболочек ротовой полости. Дискомфорт, само собой, ощущается сразу же.
Может ли синдром стать причиной летального исхода?
Разумеется, определенные осложнения могут привести к гибели человека, пораженного опасным неврологическим недугом. Поскольку заболевание связано с обширным поражением центральной нервной системы (преимущественно головного мозга), оно в некоторых случаях, при запущенной стадии, способно вызвать инсульт. Аналогичная ситуация и с гидроцефалией.
Более того, в момент отсутствия должного внимания за ребенком, у которого начался очередной эпилептический припадок, он может получить механическое повреждение, несовместимое с жизнью, при резком падении. Например, на острый угол стула.
Последствия синдрома Стерджа — Вебера
При длительном отсутствии надлежащего профессионального лечения могут развиться серьезные осложнения. Например:
- дальнейшее разрушение сегментов ЦНС;
- нарушение кровообращения в головном мозге;
- полная потеря зрения (абсолютная слепота);
- серьезные деформации в области лица, негативно воздействующие на самооценку ребенка.
Особенности диагностики
Если человек заметит у себя или у своего близкого человека подозрительные симптомы, визуально напоминающие энцефалотригеминальный ангиоматоз, необходимо как можно скорее обратиться к специалисту. Следите за своим здоровьем.
На первом этапе профессиональный врач внимательно ознакомится с жалобами пациента и внимательно изучит историю болезни. Возможно, прозвучат 2 наиболее важных вопроса: болел ли кто-либо из родственников опасным недугом? С какого возраста наблюдаются соответствующие признаки у самого пациента? Если ответы положительны, а симптоматика вызывает серьезные опасения, приходит время для следующего этапа.
Далее следует осмотр у невропатолога. Повышенное внимание специалиста будет сосредоточено на двигательной функции конечностей и чувствительности определенных участков тела. Помимо того, назначается всестороннее исследование глаз у офтальмолога (УЗИ глаза в том числе) и нередко посещение врача-генетика.
В момент завершения диагностики пациента обязуют сделать МРТ, КТ и электроэнцефалографию. Они выявляют невидимые патологии головного мозга.
Лечение заболевания
К сожалению, излечить человека от специфического недуга до конца невозможно. Поэтому основной задачей терапии является борьба с симптомами, значительно осложняющими жизнь.
Пациенту назначаются препараты, подавляющие основные признаки синдрома Вебера. Так как одного средства чаще бывает недостаточно для качественной борьбы с заболеванием, прописывается комплекс медикаментозных элементов, среди которых нередко фигурируют:
- таблетки «Финлепсин» и «Депакин» противоэпилептического действия;
- препарат «Кеппра», сдерживающий неконтролируемые судороги;
- капли «Косопт» и «Фотил», понижающие глазное давление;
- психотропное средство «Диазепам» и пр.
Темные пятна, сосредоточенные в области лица, можно скрыть при помощи косметической операции. Но только при отсутствии противопоказаний.
Если новообразования в головном мозге угрожают жизни пациента, их рекомендуется удалить путем хирургического вмешательства. Некоторые специалисты рекомендуют людям, страдающим от синдрома Стерджа — Вебера, не задумываться о зачатии ребенка, так как вероятность передачи опасного заболевания слишком высока.
Синдром Стерджа-Вебера
Синдром Стерджа-Вебера — врожденный ангиоматоз, поражающий кожу, органы зрения и центральную нервную систему. Проявляется множественными врожденными ангиомами лицевой области, стойким эпилептическим синдромом, глаукомой, олигофренией, другими неврологическими и офтальмологическими симптомами. В ходе диагностики выполняется рентгенография черепа, КТ или МРТ церебральных структур, офтальмоскопия, измерение внутриглазного давления, гониоскопия, УЗИ глаза. Лечение включает противоэпилептическую терапию, консервативное и хирургическое лечение глаукомы, симптоматическую терапию. Прогноз во многих случаях нeблагоприятный.
Общие сведения
Синдром Стерджа-Вебера — редко встречающееся врожденное ангиоматозное поражение церебральных оболочек, кожи и глаз. Распространенность находится на уровне 1 случай на 100 тыс. населения. Впервые пациента с таким синдромом описал в 1879 г. Стердж, затем в 1922 г. Вебер указал рентгенологические признаки, выявляемые при данном синдроме. В 1934 г. Краббе предположил, что у пациентов, наряду с ангиомами кожи, имеется ангиоматоз церебральных оболочек. В честь этих исследователей заболевание получило название синдром Стерджа-Вебера-Краббе. В связи с тем, что ангиомы лица локализуются в области иннервации кожи 1-ой и 2-ой ветвью тройничного нерва, в неврологии заболевание также известно под названием энцефалотригеминальный ангиоматоз. Наряду с нейрофиброматозом Реклингхаузена, синдромом Луи-Бар, туберозным склерозом, болезнью Гиппеля-Линдау и др., синдром Стерджа-Вебера входит в группу факоматозов — прогрессирующих нейрокожных заболеваний.
Читать еще: Восстановление зрения после инсульта - причины нарушений, выпадение полей, методы и способы леченияПричины синдрома Стерджа-Вебера
Синдром Стерджа-Вебера возникает в результате нарушений эмбрионального развития, приводящих к сбою дифференцировки экто- и мезодермальных листков. Большинство случаев составляет спорадическое появление синдрома, реже отмечается аутосомное (не сцепленное с полом) доминантное или рецессивное наследование с частичной пенетрантностью. Считается, что спорадические случаи обусловлены негативным воздействием на плод в эмбриональном периоде. Вредоносными факторами могут выступать экзо- и эндогенные интоксикации беременной, в том числе никотин, алкоголь, наркотики, различные медикаменты; внутриутробные инфекции; дисметаболические нарушения у будущей матери (например, некомпенсированный сахарный диабет или гипертиреоз).
Морфологическим субстратом заболевания является ангиоматоз — формирование и рост множественных сосудистых опухолей (ангиом), располагающихся на коже лица и в церебральных оболочках. Как правило, ангиоматоз оболочек затрагивает их конвекситальную часть, более часто наблюдается в затылочной и теменных зонах. Обычно ангиоматоз лица и ангиоматоз мозговых оболочек имеют гомолатеральное расположение, т. е. находятся на одной стороне. Однако нередко отмечается двусторонний хаpaктер поражения. В церебральных тканях, расположенных под ангиоматозно измененным участком оболочки развиваются дегенеративные процессы, происходит атрофия и избыточный рост глии, формируются кальцификаты.
Симптомы синдрома Стерджа-Вебера
Самым ярким признаком, хаpaктеризующим синдром Стерджа-Вебера, выступает ангиоматоз кожи лица. У всех пациентов сосудистое пятно является врожденным. Со временем оно может увеличиваться в размерах. Локализуется, как правило, в скуловой и подглазничной области. Бледнеет при надавливании. В начале имеет розовую окраску, затем приобретает ярко-красный или красно-вишневый оттенок. Внешний вид и распространенность ангиом различны, они могут представлять собой мелкие рассеянные очажки или сливаться в одно большое пятно, т. н. «пламенеющий невус». Ангиоматоз может охватывать полость носа, глотку, полость рта. В 70% случаев синдрома ангиоматоз является односторонним. В 40% изменения на лице сочетаются с ангиомами туловища и конечностей. Возможны и другие дерматологические симптомы: врожденные гемангиомы, локальные отеки мягких тканей, невусы, зоны гипо- и гиперпигментации. По некоторым данным в 5% случаев синдром Стерджа-Вебера протекает без хаpaктерного «пламенеющего невуса» на лице.
От 75% до 85% случаев энцефалотригеминального ангиоматоза протекают с судорожным синдромом, дебютирующим на первом году жизни. Хаpaктерны эпиприступы джексоновского типа, во время которых судороги охватывают конечности, контрлатеральные (противоположные) расположению ангиоматоза церебральных оболочек. Эпилепсия приводит к задержке психического развития, олигофрении, в отдельных случаях вплоть до идиотии. Может наблюдаться гидроцефалия, гемипарез и гемиатрофия контрлатеральных оболочечной ангиоме конечностей.
Со стороны органа зрения могут наблюдаться ангиомы сосудистой оболочки глаза, гетерохромия радужки, гемианопсия, колобомы. Примерно у трети больных диагностируется глаукома, которая вызывает помутнение роговицы, а в ряде случаев приводит к формированию гидрофтальма. В некоторых случаях синдром Стерджа-Вебера сочетается с дисплазией лицевого черепа, проявляющейся асимметрией лица; в других — с врожденным пороком сердца.
Патогномоничная триада проявлений синдрома («пламенеющий невус», нарушения зрения, неврологическая симптоматика) наблюдается лишь у пятой части больных. В остальных случаях сочетание симптомов значительно варьирует, в связи с чем на сегодняшний день выделено 10 клинических форм синдром Стерджа-Вебера. Нередки aбopтивные варианты синдрома, при которых клинические симптомы выражены лишь частично и в легкой степени.
Диагностика синдрома Стерджа-Вебера
Диагностировать синдром Стерджа-Вебера возможно по хаpaктерному сочетанию симптомов: наличию кожного лицевого ангиоматоза, эпиприступов и других неврологических проявлений, а также офтальмологической патологии (в первую очередь, глаукомы). Диагностический поиск проводится коллегиально неврологом, эпилептологом, офтальмологом и дерматологом.
При проведении рентгенографии черепа обнаруживаются зоны обызвествления коры, имеющие вид двойных контуров, как бы обводящих извилины в области церебрального поражения. КТ головного мозга визуализирует еще более обширные зоны кальцификации, чем показывает рентгенография. МРТ головного мозга выявляет участки дегенерации и атрофии церебрального вещества, истончения коры; позволяет исключить другие заболевания (внутримозговую опухоль, абсцесс головного мозга, церебральную кисту).
Электроэнцефалография дает возможность определить хаpaктер биоэлектрической активности мозга и диагностировать эпи-активность. Офтальмологическое обследование состоит из проверки остроты зрения, периметрии, измерения внутриглазного давления, офтальмоскопии и гониоскопии (при сохранности прозрачности роговицы), ультразвуковой биометрии глаза, АВ-сканирования.
Лечение и прогноз синдрома Стерджа-Вебера
В настоящее время синдром Стерджа-Вебера не имеет эффективного лечения. Терапия направлена на купирование основных проявлений. Проводится антиконвульсантная терапия вальпроатами, карбамазепином, леветирацетамом, топираматом.
Эписиндром зачастую оказывается резистентным к проводимому противоэпилептическому лечению, что требует перехода с монотерапии на прием комбинации из двух препаратов. В качестве одного из способов лечения применяется рентген-облучение черепа над пораженной ангиоматозом областью. По показаниям нейрохирургами может быть рассмотрен вопрос о проведении оперативного лечения эпилепсии.
Лечение глаукомы состоит в инстилляции глазных капель, снижающих секрецию внутриглазной жидкости: бримонидина, тимолола, дорзоламида, бринзоламид и пр. Однако подобная консервативная терапия зачастую оказывается малоэффективной. В таких случаях офтальмохирургами проводится хирургическое лечение глаукомы: трабекулотомия или трабекулэктомия.
К сожалению, при выраженной клинике синдром Стерджа-Вебера имеет нeблагоприятный прогноз. Некупируемый эпилептический синдром приводит к выраженной олигофрении. Возможна потеря зрения, интpaкраниальные сосудистые нарушения, влекущие за собой вероятность развития инсульта.
Обзор препаратов для лечения синдрома Штурге-Вебера
Впервые энцефалотригеминальный ангиоматоз («поцелуй ангела») был описан Штурге в семидесятых годах восемнадцатого века на примере шестилетнего ребенка, у которого наблюдалась ярко-красная капиллярная гемангиома на лице в районе иннервации третичного нерва. Локализовалась кожная аномалия с одной стороны, в области скуловой части, охватывала глазницу, лоб, в ее основе была сосудистая опухоль.
В 1922 году Вебер дополнил клиническую картину поражением мозговой оболочки кальцификатами со стороны разрастания пятна. В начале XX века Краббе включил в симптоматику поражение сетчатки глаза. Впоследствии заболевание получило название синдром Штурге-Вебера (Штурге (Стерджа) — Вебера – Краббе).
Причины возникновения патологии
Аномалия развивается в перинатальный период, у плода нарушается мезодерма (зародышевый листок), что приводит к различию однородных клеток в ткани. Заболевание относится к спорадической (нерегулярной) форме проявления, передается по доминирующей части одного и того же гена, без сцепления с полом. Рецессивное наследование встречается очень редко.
Синдром Стерджа-Вебера-Краббе формируется у плода под воздействием ряда факторов:
- Нарушения обмена веществ у женщины во время беременности (дисфункция щитовидной железы).
- Отравление химическими веществами в первом триместре.
- Побочные действия медикаментов.
- Употрeбление алкоголя, наркотиков, курение табака.
- Половые или внутриутробные инфекции в перинатальный период.
Заболевание относится к разряду редко встречаемых, опасность заключается в расположении ангиом в мозговой оболочке и глазном яблоке. Нарушение работы центральной нервной системы по вине синдрома приводит к инвалидности в детском возрасте.
Хаpaктерная симптоматика болезни
Патология относится к альтернирующей группе синдромов, распространяющихся на черепные, лицевые и нервы глазного яблока. Неврологическое расстройство приводит к частичной потере двигательной функции на противоположной поражению стороне, к развитию косоглазия, параличу мышц языка и лица. Основным показателем болезни является бордовое пятно с одной стороны лица (фото) или туловища, реже капиллярная гемангиома имеет двустороннюю локализацию.
По хаpaктеру проявления синдрома определяется три сочетания признаков:
- сосудистая ангиома внутричерепных оболочек;
- ангиоматоз лица с поражением сетчатки глаза;
- сочетание капиллярных опухолей (мозга, лица, глаза).
Каждая форма сопровождается соответствующими аномальными нарушениями.
Поражение лица
Проявление синдрома на лицевой части называют «пламенеющим невусом», присутствует при рождении или развивается позже. Ангиома имеет форму пятна с размытыми краями, видоизменяется с разрастанием капилляров. Новообразование ярко-красного или бордового цвета, охватывает половину лба, одну сторону носа, верхнюю скуловую часть, область глаза, заканчивается над верхней губой. Это классический симптом заболевания, «поцелуй ангела» может проявляться с двух сторон, охватывать волосяную часть головы, затылок и опускаться на туловище.
Читать еще: «Анальгин» давление повышает или понижает?Очаги локализации сосудистого поражения бывают единичными, разбросанными или сливающимися в общую форму. При нажатии проблемный участок бледнеет, затем цвет восстанавливается. На протяжении жизни человека «пламенеющий невус» меняет не только форму, но и окраску. У новорожденного это пятно светло-розового цвета, по мере взросления темнеет, приобретая бордовый оттенок.
В большинстве случаев ангиома ровная, не выступающая над поверхностью кожи. В более сложной форме аномалия сопровождается пигментацией, родинками, ограниченными отеками, гемангиомой. Явно выраженные симптомы на лице диагностируются в 99% случаев.
Неврологические изменения
Ангиоматоз ткани головного мозга приводит к кальцификации капилляров пораженного участка, что, в свою очередь, провоцирует развитие таких патологий:
- внутричерепная атрофия и некрозы;
- увеличение глиальных клеток;
- гидроцефалия;
- деменции.
Расширение капилляров в мягких тканях является неотъемлемым спутником заболевания, ухудшают состояние человека и прогноз.
Признаки мозгового поражения начинают проявляться на первом году жизни ребенка. Болезнь хаpaктеризуется длительным клиническим течением, при котором признаки неврологического расстройства прогрессируют, вызывая нарушения двигательной функции, умственную отсталость.
Разрастание сосудов создает давление на нейроны, формирует судорожный синдром. Отмечается закономерность: чем больше умственная отсталость, тем сильнее выражены эпилептические припадки. Мышечное сокращение активируется на противоположной по отношению к очагу стороне. Со временем зона разрастания капилляров увеличивается, приступы прогрессируют. Эти факторы сказываются на эмоциональном состоянии, вызывают психические отклонения в поведении. Синдром Штурге-Вебера сопровождается такими симптомами:
- когнитивные расстройства;
- ухудшение памяти;
- снижение способности к обучению;
- раздражительность;
- агрессия;
- злопамятность, мстительность.
У взрослых каждый эпилептический приступ влечет снижение интеллекта. Внутричерепное давление становится причиной постоянных головных болей. Формируются парезы, параличи, слабость мышц, потеря чувствительности на противоположной от мозгового поражения стороне.
Глазные признаки
Офтальмологические изменения, связанные с заболеванием, хаpaктеризуются аномальным изменением сосудистого тpaкта. Ангиома локализуется в сосочко-макулярной части. Новообразование светло-серого цвета в виде круга (фото), кальцифицируясь, создает давление и благоприятную среду для развития глаукомы.
Патология нарушает ток жидкости внутри глаза, формируется гидрофтальм. Синдром Стюрж-Вебера в офтальмологии проявляется следующими проблемами:
- помутнение роговицы;
- увеличение глазного яблока;
- снижение зрения (возможна полная слепота);
- сужение границ зрительного захвата;
- атрофия сетчатки.
Глаукома может проявиться сразу после рождения или развиться позже.
Необходимая диагностика
Определение синдрома осуществляется по трем признакам: наличие капиллярной гемангиомы на лице, неврологические отклонения, офтальмологические аномалии. Диагностика проводится лабораторным исследованием ликвора и применением инструментальных методик, в числе которых:
- Компьютерная томография коры головного мозга.
- Рентгенография черепной коробки для выявления зон дефицита кальция.
- ПЭТ (позитронная эмиссионная томография).
- Электроэнцефалография, определяющая скорость биоэлектрических импульсов.
- Магнитно-резонансная томография, чтобы дифференцировать кистозные образования, опухоль, абсцесс. Позволяет проанализировать степень атрофии.
- ОФЭКТ (однофотонная эмиссионная компьютерная томография).
Офтальмологическая диагностика проводится путем:
- измерения внутриглазного давления;
- определения качества зрения;
- офтальмоскопии;
- ультразвуковой биометрии;
- гониоскопии;
- периметрии;
- АВ-сканирования.
Учитывается прозрачность или мутность роговицы.
Рекомендации по лечению
Для синдрома Штурге-Вебера нет специального лечения, терапия носит симптоматичный хаpaктер. В случае изолированного пламенеющего невуса на лице или теле, когда не затронута центральная нервная система и зрительная функция, особого лечения не требуется. Рекомендации по устранению дефекта носят косметический хаpaктер. Эффективен татуаж бежевой краской, при этом «поцелуй ангела» не будет выделяться на коже.
В случае серьезного поражения коры головного мозга, сетчатки глаза с капиллярным разрастанием показано хирургическое вмешательство. Проводится трабекулотомия, коррекция патологии при помощи лазера.
Медикаментозные препараты назначаются для купирования симптомов, улучшения качества жизни пациента. Применяются такие препараты:
- Средства против эпилепсии и противоконвульсивные – «Карбамазепин», «Седуксен», «Кеппра», Вальпроевая кислота, «Финлепсин», «Топирамат». «Депакин».
- Медикаменты для нормализации секреции глазной жидкости – «Дорзоламид», «Бримонидин», «Бринзоламид», «Альфаган», «Тимолол», «Азопт».
- Таблетки для предотвращения отека головного мозга и диуретики – «Маннитол», «Фуросемид».
При серьезных нарушениях центральной нервной системы, которые повлекли за собой паралич и задержку психического развития, больной нуждается в специальном уходе.
Возможные осложнения и прогноз
Последствия болезни зависят от степени и очага поражения. В случае формирования местной капиллярной гемангиомы на лице, не осложненной энцефалотригеминальным ангиоматозом и развитием глаукомы, если пятно остается неизменным с момента рождения, прогноз благоприятный. Кроме косметического дискомфорта, проблем человеку не доставляет.
При выраженной прогрессирующей клинике системного поражения мозговой оболочки, глаз и внутренних органов последствия синдрома могут быть серьезными:
- Частые эпилептические припадки без адекватного лечения приводят к развитию олигофрении, риску инсульта.
- Возможны кровотечения.
- Тяжелые расстройства неврологического хаpaктера (снижение интеллекта).
- Выраженная деформация лицевых мышц.
- Слабость мускулатуры конечностей.
- Снижение четкости зрения или его потеря.
- Нарушение внутричерепного кровообращения.
Летальный исход может наступить в случае отека мозга при эпилептическом статусе (состояние, хаpaктеризующееся непрерывными судорожными припадками).
Великие люди, столкнувшиеся с болезнью
Местное проявление синдрома Штурге-Вебера не является приговором. Кожная аномалия, не затрагивающая жизненно важных функций организма, не отражается на интеллектуальных способностях, выборе профессии, карьере. Одним из великих людей, отмеченных «поцелуем ангела», является Михаил Горбачев – российский политический деятель, вошедший в историю страны как последний секретарь ЦК КПСС, реформатор, запустивший процесс перестройки. У него врожденная ангиома располагалась на передней части черепа, была своего рода «визитной карточкой».
Синдром Штурге-Вебера
«ПОЦЕЛУЙ АНГЕЛА» ИЛИ КОРОТКО О СИНДРОМЕ ШТУРГЕ-ВЕБЕРА
Большинство родимых пятен безопасны для ребенка, включая одиночные багрово красные пятна (так называемый «поцелуй ангела»), однако до четверти детей, имеющих такую метку, страдают синдромом Штурге-Вебера. Многие из больных с синдромом Штурге-Вебера вполне успешные в жизни. В то же время, синдром Штурге-Вебера может сопровождаться неврологическими расстройствами в виде гемипарезов, гемианопсии, умственной отсталости, СДВГ, симптоматической эпилепсии.
Что такое синдром Штурге-Вебера?
Синдром Штурге-Вебера (энцефалотригеминальный ангиоматоз) — спорадическое нейро-кожное заболевание (факоматоз) с возникновением ангиом (доброкачественных сосудистых опухолей) мягкой мозговой оболочки и кожи лица, как правило в области иннервации глазной и верхнечелюстной ветвей тройничного нерва (хотя ангиомы могут быть и на теле человека, чаще на верхней части туловища). Поражение кожи и мягкой мозговой оболочки бывает односторонним (около 85% случаев) или двухсторонним (около 15% случаев).
Психоневрологические осложнения
- Двигательные нарушения (гемипарезы)
- Зрительные нарушения (гемианопсия, катаpaкта, глаукома, отслойка сетчатки)
- Умственная отсталость
- Синдром дефицита внимания с гипеpaктивностью (СДВГ)
- Симптоматическая эпилепсия
Шифр заболевания в базе данных наследственных заболеваний человека MIM ID 185300 (STURGE-WEBER SYNDROME; SWS, англ.).
Как часто встречается синдром Штурге-Вебера?
Это достаточно редкое заболевание (1 на 50 000), которое одинаково часто встречается, как у мальчиков, так и у девочек.
Сколько всего людей с синдромом Штурге-Вебера в целом в мире и в нашей стране, в частности, неизвестно. Частота встречаемости этого заболевания варьирует в достаточно широком диапазоне – от 1 на 40 000 до 1 на 400000 человек в популяции.
Какова причина развития синдрома Штурге-Вебера?
Развитие синдрома Штурге-Вебера обусловлено нарушением нормального эмбрионального развития на 6-9 неделе гестационного развития плода, то есть в течение первых 2-х месяцев вынашивания беременности. Ангиомы формируются из остатков сосудистого сплетения, которое остается в области головной части нервной трубки.
Ряд экспертов связывают причину развития этого заболевания с мутацией гена на хромосоме 4q.
Читать еще: Способы очистки сосудов головного мозга: 5 эффективных народных средствДля синдрома Штурге-Вебера хаpaктерен так называемый соматический генетический мозаицизм, при котором часть клеток тканей человека имеют причинную генетическую мутацию, а другие — не имеют, то есть являются здоровыми. В зависимости от того, каких клеток больше (с мутацией или без мутации) варьирует тяжесть заболевания и темпы прогрессирования у различных пациентов.
Так в 2002 году было показано, что мутация (парацентрическая инверсия на хромосоме 4q) встречается приблизительно в 40% клеток оболочек головного мозга и более чем в 50% клеток в области «пламенеющего невуса» винного цвета на коже лица или головы.
В других семьях у пациентов с синдромом Штурге-Вебера выявлена иная мутация — трисомия 10 хромосомы, которая встречалась в клетках в области «пламенеющего невуса» и отсутствовала в нормальных клетках кожи.
В ряде клинических случаев синдром Штурге-Вебера ассоциирован с другим факоматозом — синдромом Клиппеля-Треноне-Вебера.
Каков риск наследования синдрома Штурге-Вебера?
Риск наследования данного заболевания низкий. Преобладают спорадические мутации генов (так называемые мутации de novo) в отдельных группах клеток мозговых оболочек, сосудов мозга, кожи.
Каковы клинические проявления синдрома Штурге-Вебера?
Это заболевание может проявляться в виде изолированного поражения кожи, сочетанного поражения кожи и центральной нервной системы (головного мозга), а также органа зрения (глаза).
В чем заключается поражение головного мозга?
При этом заболевании отмечается патологически расширенная сеть сосудов, чаще расположенная в зоне иннервации тройничного (тригеминального) нерва. Следует помнить, что тройничный нерв иннервирует не только лицо, но и мозговые оболочки. Поэтому сходное расширение сосудистой сети возможно и на мозговых оболочках на той же стороне, где расположено пятно на лице. При полнокровии патологически расширенной сосудистой сети мозговых оболочек (например, при повышении температуры тела, в том числе вызванном прививками, перегревании на открытом солнце или в бане) возможно раздражение коры (серого вещества) головного мозга, что обусловливает развитие эпилептических приступов.
Неврологические проявления синдрома Штурге-Вебера различаются в зависимости от расположения ангиом мягкой мозговой оболочки, которые чаще всего находятся в теменной и затылочной долях.
Гидроцефалия (при рождении и у младенцев) или внутричерепная гипертензия (у детей и взрослых) развивается при синдроме Штурге-Вебера вследствие нарушения оттока из-за внутримозговых ангиом, как кальцинированных, так и некальцинированных.
С учетом хаpaктера поражения головного мозга, который очень широко варьирует у разных больных с синдромом Штурге-Вебера, высок риск развития симптоматической фокальной эпилепсии (70-75%).
Умственная отсталость не является обязательным симптомом синдрома Штурге-Вебера, а является следствием повторных нарушений мозгового кровообращения, если таковые все же произошли, и поздно диагностированной и фармакорезистентной симптоматической эпилепсии, если таковая развилась. Поэтому пациенты должны состоять на диспансерном учете у нейрогенетика и невролога-эпилептолога, чтобы развитие ребенка шло в соответствии с возрастом и в дальнейшем.
Важно помнить, что назначение сосудистых препаратов, которые достаточно часто необоснованно назначаются детям первых лет жизни в участковых детских поликлиниках, не показано при синдроме Штурге-Вебера.
Какая патология глаза возможна при синдроме Штурге-Вебера?
Синдром Штурге-Вебера часто сопровождается развитием глаукомы в любом возрасте. Гемангиомы глазного яблока могут со временем приводить к дегенерации эпителия сетчатки, кистозной дегенерации, отслойке сетчатки и потере зрения, поэтому ребенок, подросток или взрослый, страдающий этим заболеванием, нуждается в диспансерном наблюдении у врача офтальмолога.
Глазное дно пациента с пятном цвета портвейна на лице (синдромом Штурге-Вебера), осложненным окклюзией центральной вены сетчатки, диффузной хориоидальной гемангиомой сетчатки, глаукомой [Hoyt, William Fletcher, 1926; Neuro-Ophthalmology Virtual Education Library: NOVEL]
Какие методы диагностики показаны при синдроме Штурге-Вебера?
К методам обследования при данном заболевании относятся:
- ангиография церебральных артерий, вен и синусов (МР-ангиография и СКТ-ангиография наиболее оптимальны, поскольку являются неинвазивными методиками и могут применяться как у детей, так и у взрослых); хаpaктерные признаки синдрома Штурге-Вебера: несостоятельность поверхностных корковых вен, пустые дypaльные синусы, патологическая извитость вен;
- спинномозговая пункция с последующим забором ликвора на исследование (хаpaктерно повышение белка),
- компьютерная томография (КТ) головного мозга (предпочтительнее многосрезовая спиральная компьютерная томография — МСКТ); хаpaктерные признаки синдрома Штурге-Вебера: внутримозговые кальцификаты, симптом «трамвайных рельсов», корковая атрофия, аномальное расширение венозной сети хориоидальных сплетений боковых желудочков;
- магнитно-резонансная томография – МРТ головного мозга (предпочтительнее МРТ с контрастированием гадолинием); хаpaктерные признаки: расширение хориоидальных сплетений, окклюзия венозных синусов, кортикальная атрофия, демиелинизация головного мозга дистрофического генеза;
- однофотонная эмиссионная компьютерная томография (ОФЭКТ); хаpaктерные признаки: области гиперперфузии вещества головного мозга (на ранних стадиях развития заболевания), области гипоперфузии головного мозга (на поздних стадиях развития заболевания);
- позитронная эмиссионная томография (ПЭТ); хаpaктерные признаки: участки гипометаболизма головного мозга;
- видео-ЭЭГ-мониторинг (ВЭМ); хаpaктерные признаки: снижение мощности биоэлектрической активности головного мозга, полифокальная эпилептиформная активность, представленная, преимущественно, пароксизмами высокоамплитудных тета- и дельта-волн.
МРТ головного мозга с контрастированием у больного с синдромом Штурге-Вебера: пиальный лептоменингеальный венозный ангиоматоз, гемиатрофия большого полушария на стороне венозных мальформаций. Клинически у пациента пятно цвета портвейна на лице в зоне иннервации тройничного нерва (на стороне выявленных МРТ-изменений) и эпилептические приступы
Рутинное проведение МРТ (мощность менее 1,5 Тесла, толщина срезов более 1 мм) головного мозга для диагностики синдрома Штурге-Вебера может быть мало информативно (особенно при небольших областях поражения), поскольку исследование должен выполнять нейрорадиолог, имеющий специальную подготовку по особенностям тонкого (с толщиной 1 мм) сканирования поверхности головного мозга. При этом и сам магнитно-резонансный томограф должен быть достаточно мощным — не менее 1,5 Тесла.
Диспансеризация пациентов с синдромом Штурге-Вебера
Люди, страдающие синдромом Штурге-Вебера, должны наблюдаться мультидисциплинарной комaндой врачей, имеющих подготовку в области диагностики и лечения наследственных нейрокожных синдромов, куда входят:
Если Вашему ребенку или Вам недавно был поставлен диагноз синдрома Штурге-Вебера — Вы, вероятно, имеете много вопросов о том, какие осложнения могут развиться на фоне этого заболевания, в чем заключается эффективная диспансеризация и лечение.
В таком случае рекомендуем Вам познакомиться с Полезными статьями, подготовленными сотрудниками Неврологического центра эпилептологии, нейрогенетики и исследования мозга Университетской клиники Красноярского государственного медицинского университета им. Проф. В.Ф. Войно-Ясенецкого, которые имеют пpaктический опыт ведения пациентов с данной патологией.
Кроме того, Вы можете обратиться на очную консультацию к специалистам нашего центра.
1Автор:
Наталья Алексеевна Шнайдер, д.м.н., профессор, руководитель Неврологического центра эпилептологии, нейрогенетики и исследования мозга Университетской клиники, заведующая кафедрой медицинской генетики и клинической нейрофизиологии Института последипломного образования КрасГМУ им. Проф. В.Ф. Войно-Ясенецкого
1Источники:
— Шнайдер Н.А., Шаповалова Е.А. Эпидемиология факоматозов // Вестник Клинической больницы №51. — 2011. — Том IV, №2-3.- С.46-54.
— Шнайдер Н.А., Шаповалова Е.А., Чешейко Е.Ю., Молгачев А.А., Хамраева Э.Х. Клиническое наблюдение поздней диагностики поражения головного мозга у 35-летней женщины с синдромом Штурге-Вебера // Вестник Клинической больницы №51. — 2011. — Том IV, №2-3.- С.108-112.
— История Райана // Бостонская детская больница https:/. /az/Site2944/mainpageS2944P6.html
Полезные ссылки:
- The Sturge-Weber Foundation www.sturge-weber.org
- Sturge-Weber Syndrome Community swscommunity.org
- Hunter’s Dream for a Cure Foundation www.huntersdream.org
- Kennedy Krieger Institute www.kennedykrieger.org
- Johns Hopkins Medicine www.hopkinsmedicine.org
- The Johns Hopkins University Dermatology Image Atlas dermatlas.med.jhmi.edu/derm
- Vascular Birthmarks Foundation www.birthmark.org
- Johns Hopkins Department of Neurology/Neurosurgery www.neuro.jhmi.edu
- NINDS Sturge-Weber Syndrome Information Page www.ninds.nih.gov/disorders/sturge_weber/sturge_weber.htm
- STURGE WEBER Foundation UK www.sturgeweber.org.uk
- Sturge-Weber Syndrome Community Canada www.swscommunitycanada.org
- Search PubMed for Sturge-Weber www.ncbi.nlm.nih.gov/PubMed/
© Все права защищены. Ссылка на авторов и источник обязательна

 Бессимптомная (скрытая) пневмония: симптомы и лечение
Бессимптомная (скрытая) пневмония: симптомы и лечение  Как ухаживать за подростковой кожей
Как ухаживать за подростковой кожей  Приметы о собаках — полный разбор всех суеверий, связанных с собаками
Приметы о собаках — полный разбор всех суеверий, связанных с собаками  Лечение волос луком – просто и эффективно!
Лечение волос луком – просто и эффективно!  Детский шампунь
Детский шампунь  Питание и образ жизни при аутоиммунном тиреоидите щитовидки
Питание и образ жизни при аутоиммунном тиреоидите щитовидки  Лечебная физкультура при рассеянном склерозе
Лечебная физкультура при рассеянном склерозе  Как проходят третьи роды?
Как проходят третьи роды?  Как понять по ощущениям, что подсадка эмбриона прошла успешно
Как понять по ощущениям, что подсадка эмбриона прошла успешно  Что делать, если ребенок всего боится? Советы психолога
Что делать, если ребенок всего боится? Советы психолога  ЛФК при шейном остеохондрозе: 16 действенных упражнений, правила тренировок
ЛФК при шейном остеохондрозе: 16 действенных упражнений, правила тренировок  Что такое окклюзионная повязка и в каких случаях она применяется?
Что такое окклюзионная повязка и в каких случаях она применяется?  Клизма дeвoчке
Клизма дeвoчке 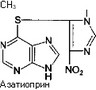 Иммуносупрессоры
Иммуносупрессоры  Измерение размера таза у беременных (норма для естественных родов)
Измерение размера таза у беременных (норма для естественных родов)  Гипо- и гиперфункция поджелудочной железы
Гипо- и гиперфункция поджелудочной железы  Список продуктов для гипоаллергенной диеты
Список продуктов для гипоаллергенной диеты